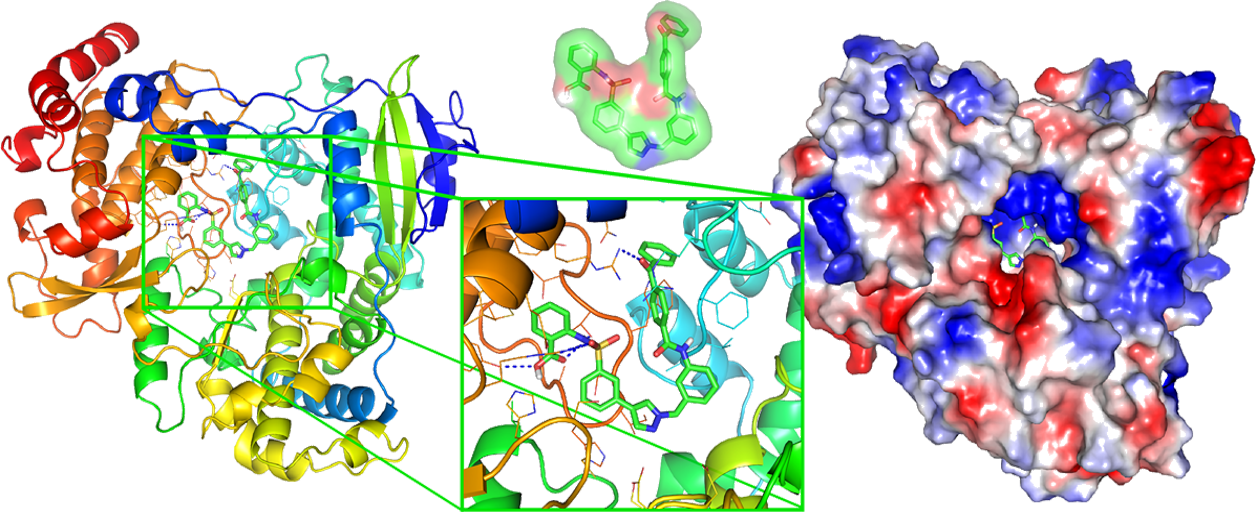
Corrosion inhibition from first principles: the role of the oxide
- Investigador principal: Pablo Ordejón.
- Investigadores: Alberto García Arribas, José María Castillo Robles, Pol Febrer, Roberta Farris, José María Escartín, Vladimir Dikan, Irina Lebedeva, Federico Pedrón, Catalina Coll.
Corrosion is a critical technological problem, and finding green alternatives to the toxic inorganic compounds used to prevent it is a very hot research topic. The use of small organic molecules as inhibitors has increased significantly in recent years due to their low toxicity. Nevertheless, the mechanisms underlying corrosion inhibition (CI) by these molecules are still unknown.
Molecular modelling was widely applied to find structure-activity relationships for several inhibitors. However, the CI process is a complex problem that requires more refined simulations to be properly understood.
Here we propose using a state-of-the-art methodology combining DFT, NEGF, and QM/MM to tackle that problem, allowing us to reach long MD simulations fully accounting for the potential applied to the electrodes and the solvent effects.